Upiorna hekatomba krów, która przetoczyła się przez świat w ostatnich latach ubiegłego wieku nie wywołała dostatecznego wrażenia, a przecież masowa rzeź bydła dotkniętego „chorobą wściekłych krów” powinna nam dać do myślenia. Ilekroć bowiem człowiek zaczyna zabawiać się w Stwórcę realizując plany powszechnej szczęśliwości, tylekroć potyka się o własne nogi i przewraca. Ta „utylizacja chorych zwierząt” w istocie może się kojarzyć z pogańskimi ofiarami całopalnymi, składanymi dla przebłagania rozgniewanych bóstw. Zapłaciliśmy cenę za pełne pychy przekonanie, że możemy ulepszyć naturę czyniąc z hodowli bydła proces przemysłowy, gdzie kryteria ekonomiczne dopuszczają działania, których by się powstydził każdy gospodarz w swojej oborze. Chodzi oczywiście o paszę, w której krowy zjadały mączkę ze swoich padłych koleżanek.
Nieprzewidywalne priony
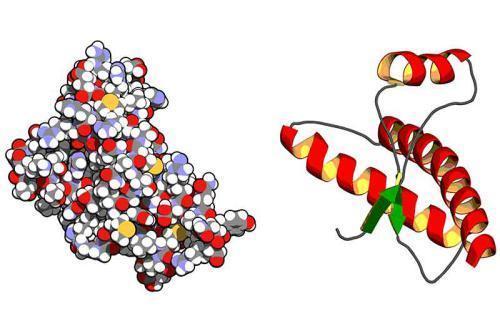
„Choroba wściekłych krów”, czyli BSE (encefalopatia gąbczasta bydła) jak i pokrewna „trzęsawka” na którą zapadają owce, kozy i muflony, początkowo wydawała się nieszkodliwa dla człowieka. Mimo to interesowali się nią nie tylko weterynarze. Już w roku 1984 Stanley B. Prusiner (późniejszy noblista), wysunął przypuszczenie, że czynnikiem chorobotwórczym jest specjalny rodzaj białka, które nazwał prionowym. Hipoteza ta była dość rewolucyjna, gdyż negowała powszechnie przyjęty pogląd, że patogeny muszą zawierać DNA lub RNA, aby reprodukować się w zaatakowanym organizmie. Dziś wiemy, że istotnie priony są odpowiedzialne za BSE i kilka innych encefalopatii, włączając ludzkie choroby: Creutzfeldta-Jakoba, Gerstmanna-Strausslera-Scheinkera, choroby „kuru” (o czym dalej) oraz tzw. „śmiertelnej bezsenności”. Ta ostatnia to choroba mózgu dziedziczona w 50 % przypadków. Odpowiedzialny za nią gen został wykryty u zaledwie 28 rodzin na świecie. Istotą choroby jest całkowita niezdolność do snu, która zawsze jest nieuleczalna i prowadzi do śmierci.
Jednak nie „dziwność” tych chorób stanowiła największe zaskoczenie. Badaczy bardziej zadziwił fakt istnienia „normalnej” formy prionu (zwanej komórkową), stymulującej działanie komórek nerwowych. Pojawiające się formy chorobotwórcze tego białka są na bieżąco usuwane. Kiedy jednak ten mechanizm zawodzi, wadliwe priony rozmnażają się lawinowo, jak cząsteczki w termojądrowej reakcji łańcuchowej. Potem wytwarzają skupiska (beta amyloidy) odporne na wysoką temperaturę, promieniowanie i związki chemiczne, które wkomponowując się w ubytki masy mózgowej tworzą strukturę podobną do gąbki. Choroby prionowe, jak się potem okazało, mogą powstawać w wyniku zjedzenia zakażonej wołowiny, jednak mogą pojawiać się całkiem spontanicznie (w przypadku Creutzfeldta-Jakoba 1 osoba na milion), lub przez mutację genu wytwarzającego priony (jedna osoba na 10 mln). Ta pierwsza możliwość budziła kontrowersje i ten wariant choroby Creutzfeldta-Jakoba bywał traktowany jako oddzielna jednostka chorobowa. W tych przypadkach zachodziły też procesy gromadzenia się „złych”prionów w innych narządach (np. układzie limfatycznym), co oznacza, że zostały przeniesione za pośrednictwem krwi. Udowodniono śmiertelne przypadki w zakażeniach tą drogą, toteż na przykład osoby urodzone po 1996 (kiedy to wreszcie zaprzestano przymuszać krowy do kanibalizmu) są w Wielkiej Brytanii chronione przed przetaczaniem krwi od swoich ziomków urodzonych wcześniej. W USA z kolei dawcami krwi nie mogą być osoby przebywające w Zjednoczonym Królestwie ponad 3 miesiące. Istnieją przeciwnicy rozbudowywania olbrzymiego aparatu diagnostycznego i systemu prewencji (my „na szczęście” nie musimy obawiać się nadgorliwości służb medycznych), bowiem ilość przypadków śmiertelnych klasyfikuje choroby wywołane prionami na odległych miejscach statystyk. Jednak ze względu na długi okres utajenia choroby, wielość szczepów w jakich występują „złe” priony, a przede wszystkim fakt istnienia spontanicznych zachorowań (tak u ludzi jak u zwierząt) nie związanych z jedzeniem ani mutacją genu, mamy prawo traktować przyszłość tych chorób jako groźną niewiadomą. Zresztą badania związane z profilaktyką o diagnozowaniem dostarczają coraz to nowych danych o biologii prionów, co na znaczenie dla rozwoju nauki jak i praktyki medycznej. Kardynalnym bowiem błędem jest traktowanie chorób generowanych prionami na podobieństwo wirusowych epidemii. Podobieństwa są duże, lecz mechanizmy działania prionów są inne; jakie – o tym przekonujemy się wciąż na nowo.
Dygresja kulinarna
Na własnej skórze odczuli to członkowie plemienia Fore na Nowej Gwinei, których przetrzebiła choroba prionowa o nazwie „kuru”, co oznacza „śmiejącą się śmierć”. Prawdopodobnie jeden z członków plemienia zapadł na nią spontanicznie, za to już zupełnie konwencjonalnie zachowali się inni dżentelmeni, a mianowicie spożyli mózg denata w trakcie obrzędu grzebalnego, jak nakazuje tradycja. Kobiety i dzieci poprzestawały na nacieraniu się potrawą. Pragnęli w ten sposób przyswoić sobie mądrość i doświadczenie czcigodnego zmarłego, a tymczasem natrafili na podstępne priony, które dały znać o sobie dopiero po 20 latach. Nie wiadomo, czy to zjedzony przodek okazał się także nośnikiem uzdrawiającej wiedzy, w każdym razie 1959 roku odstąpiono do kanibalistycznych przekąsek i epidemia ustała.
Wiara w to, że przez zjedzenie przodka można przejąć jakieś jego umiejętności nie jest tak bzdurna, jak się początkowo wydaje. Przypominają się w tym miejscu eksperymenty na szczurach z lat 60., kiedy jeszcze nie znano prionów. Mózgi doświadczonych w trudnej sztuce poruszania się po labiryncie zwierząt służyły jako pokarm młodszej generacji. I okazywało się, że niedoświadczone osobniki znały sposoby unikania kar i przejmowania nagród, chociaż nie miały okazji nauczyć się tego metodą prób i błędów. Te wyniki były nieco kłopotliwe, gdyż niejako przeczyły teorii ewolucji. Cechy nabyte nie podlegają przekazywaniu następnym pokoleniom. Jak widać czego nie można przekazać przez geny, można zjeść.
Trzeba też wspomnieć o roli prionopodobnych białek w takich na przykład procesach jak fermentacja drożdżowa. Całe pokolenia miłośników wina i piwa jeśli miały na ten temat jakieś wyobrażenia, były to podręcznikowe rysunki komórek przenoszących swe cechy na potomstwo przy pomocy nici DNA. Tymczasem priony potrafią wytłaczać swe podobizny bez pośrednictwa kodu genetycznego. Ściśle mówiąc, zmiany fenotypu komórki drożdżowej przekazują jakąś informację genetyczną, chociaż bez udziału kwasów nukleinowych.
Żmudne poszukiwania
W trakcie prac nad testami wykrywającymi „złe” priony badano odporność poszczególnych form prionowych na proteazę, enzym rozkładający białka. Okazało się, że stopień odporności jest różny i prawdopodobnie wszystkie priony są początkowo wrażliwe na proteazę. Okazało się również, że lokalizacja prionów może także, prócz mózgu, obejmować różne grupy mięśni. Ponadto znaleziono ponad 20 substancji hamujących rozwój prionów, chociaż niektóre z nich są zbyt toksyczne. Szczególnie godne uwagi są związki przekraczające barierę krew–mózg. Często są to środki używane w zupełnie innych dolegliwościach. Można tu wymienić chloropromazynę stosowaną od dawna w psychiatrii lub antymalaryczną atebrynę. Nie niszczą one niestety „złych” prionów, ale spowolniają ich rozwój. Innym testowanym środkiem jest polisiarczan pentozanu. Niepowodzenia nie powinny zrażać, gdyż stawka jest wysoka. Rozpracowanie obyczajów wrogich struktur białkowych leży także w interesie cierpiących na chorobę Alzheimera czy Parkinsona.
Ze względu na wyjątkową odporność na temperaturę i urazy mechaniczne, znaleziono także zastosowanie technologiczne dla prionów. Ich ultracienkie nici mają być, po powleczeniu złotem, używane jako misterne sieci przewodów elektrycznych w niektórych urządzeniach elektronicznych.
Aleksander Straszewski